MAPK-driven epithelial cell plasticity underlies CRC therapeutic resistance
(White et al., Cancer Research UK Scotland Institute and collaborators across the MRC National Mouse Genetics Network – Cancer Cluster)
Colorectal cancer (CRC) is a genetically complex disease, yet resistance to targeted therapies remains a major clinical problem, leading to unfavourable outcomes for patients. This study, recently published in Nature, and featuring significant contributions from members of the MRC National Mouse Genetics Network’s Cancer Cluster, uncovers how epithelial cell plasticity, driven by and responding to MAPK (mitogen-activated protein kinase) signalling, underlies therapeutic resistance in CRC.
Key discovery
CRC cells can switch between two distinct stem-like states:
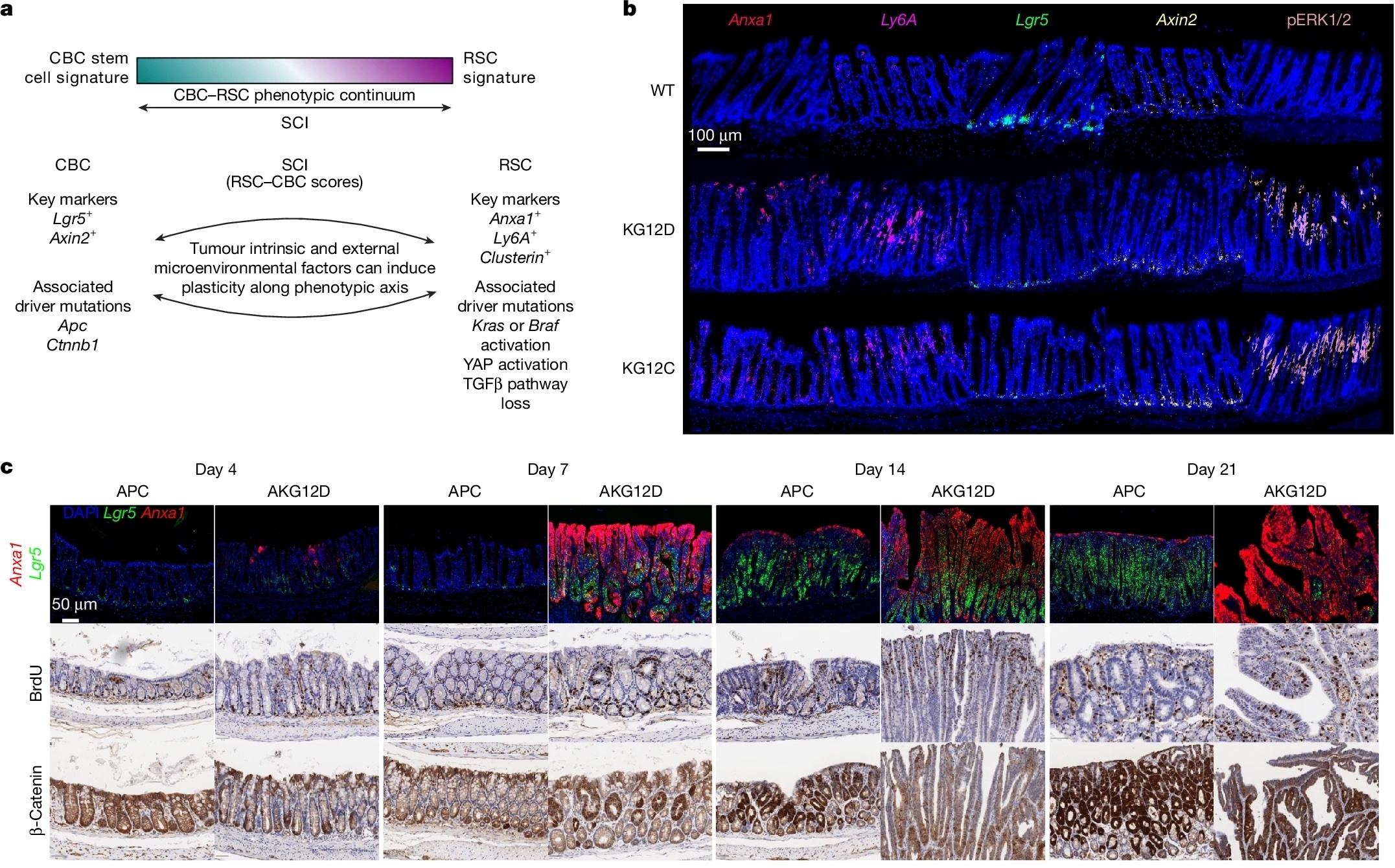
- Canonical Wnt-high stem cells (LGR5⁺ crypt-base cells), which maintain homeostatic renewal, and
- Regenerative or injury-like stem cells (Anxa1⁺, Ly6a⁺), which emerge during tissue repair or oncogenic stress.
Mutations that activate the MAPK pathway, particularly in KRAS and BRAF, push epithelial cells toward this regenerative identity. When MAPK signalling is pharmacologically inhibited, tumour cells revert towards a Wnt-dominant state instead of dying. This reversible “state-switching” allows continued growth despite effective target suppression, providing a mechanistic explanation for the transient and variable responses seen in patients treated with KRAS or BRAF inhibitors.
Methodology and models
Using an integrated platform of genetically engineered mouse models (GEMMs), tumour organoids, single-cell RNA sequencing, and spatial transcriptomics, the authors mapped how oncogenic mutations and drug treatments rewire epithelial identity in vivo. The study leveraged sophisticated models developed within the MRC National Mouse Genetics Network Cancer Cluster, demonstrating the power of national collaborative infrastructure for mechanistic cancer biology.
Major findings
- Oncogenic MAPK activation (via KRAS or BRAF mutations) drives intestinal epithelia into a regenerative, injury-associated state.
- MAPK inhibition reverses this, inducing a Wnt-high stem-like state and maintaining tumour viability, showing that resistance is adaptive and phenotypic, not purely genetic.
- Early metastases are transcriptionally uniform and sensitive to MAPK inhibitors, but as heterogeneity develops, resistance emerges—revealing a potential therapeutic window for early intervention.
- BRAF-mutant CRCs respond transiently to BRAF+EGFR inhibition because they adaptively activate Wnt signaling; combining this treatment with radiotherapy improves efficacy.
- Loss of RNF43, a negative Wnt regulator, restricts plasticity and produces “super-responders” to BRAF+EGFR therapy. This finding explains clinical observations of exceptional responses in BRAF/RNF43 co-mutant CRC patients.
Significance
This work reframes CRC drug resistance as a problem of cell identity rather than solely of mutational escape. Therapeutic success depends on controlling epithelial plasticity, either by timing interventions when tumours are least heterogeneous (e.g., during early metastasis) or by constraining fate switching (e.g., in patients with RNF43-mutant disease).
MRC Network impact
By combining advanced genetic modelling, single-cell technologies, sophisticated computational analysis, and translational insight, this research exemplifies the MRC National Mouse Genetics Network’s Cancer Cluster mission: to connect precision mouse models with clinically relevant cancer biology. The collaborative framework enabled integration of expertise from Glasgow, Oxford, and other UK partners, directly advancing understanding of how dynamic epithelial states determine CRC therapeutic outcomes.
In essence, controlling, not just targeting, cellular plasticity may be the key to durable responses in colorectal cancer.
Mark White (first author) on the scientific significance
“This study shows that resistance to targeted therapies in colorectal cancer is not simply a consequence of new mutations emerging, but of cancer cells changing who they are. We found that tumour cells can dynamically switch between distinct stem-like states in response to treatment, allowing them to survive even when key oncogenic pathways are effectively inhibited. Understanding this plasticity gives us a new framework for thinking about resistance, and importantly, points to new opportunities to intervene before or during these adaptive transitions.”
Owen Sansom (senior author and Network Director) on the contribution of the NMGN and Cancer Cluster
“This work is a strong example of what the MRC National Mouse Genetics Network, and particularly the Cancer Cluster, was established to enable. By bringing together advanced in vivo mouse models with emerging new approach methodologies, we can begin to tackle the real biological complexity that underlies therapy response and resistance. No single model or technology can capture this alone. Progress depends on integrating systems, disciplines and expertise to better reflect how disease behaves in patients.”
Philip Dunne (co-author and Cancer Cluster member) on the integration of in vivo models and computational analysis
“A major strength of this study lies in the integration of sophisticated in vivo models with advanced computational and single-cell analytical approaches. By combining spatial and single-cell transcriptomics with robust biological modelling, we can interpret tumour behaviour in far greater depth and with much clearer relevance to human disease. This kind of integrative analysis is essential for translating complex experimental data into insights that genuinely inform patient biology and clinical decision-making.”